The systematic approach developed to assess the amount of residues left on manufacturing equipment surfaces from product carryover is known as cleaning validation. Current trends have seen increasing demand for rapid sample analysis time along with low detection limits for verification of cleaning validation samples. A total organic carbon (TOC) method is sensitive to the ppb range and is less time consuming than high performance liquid chromatography (HPLC). The purpose of this study is to demonstrate how to develop and validate a TOC method for cleaning applications. Validation of the cleaning procedures for manufacturing or processing equipment has been presented in this paper. A sensitive and reproducible method was developed and validated for the determination of cephradine in swab samples. The method for determining residues of cephradine on manufacturing equipment surfaces was validated for precision, linearity, accuracy, limit of quantification and % recovery of a potential contaminant. The sampling procedure using cotton swabs was also validated. A mean recovery from stainless steel plate close to 78% was obtained. The assay was linear over the concentration range of 30 to 600 ng ml−1 concentration (R 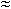 0.9987). The calculated limit of contamination value was less than 315
0.9987). The calculated limit of contamination value was less than 315 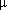 g cm−2, during three consecutive cleaning trials.
g cm−2, during three consecutive cleaning trials.
0.9987). The calculated limit of contamination value was less than 315 g cm−2, during three consecutive cleaning trials.1. Introduction
Pharmaceutical products are very much susceptible to contamination from shared manufacturing equipment. In many cases, the same equipment may be used for processing different products.1 Cleaning validation is the process of assuring that cleaning procedures effectively remove residue from manufacturing equipment below a predetermined level. This is necessary to assure the quality of upcoming product using the same equipment, to prevent cross-contamination. Good manufacturing practice in pharmaceutical manufacturing plants states that the equipment must be maintained in a clean and orderly manner.2–8 Mostly, cleaning validation samples have been measured using high performance liquid chromatography (HPLC) methods, which are often time consuming and subject to a number of interferences. Total organic carbon (TOC) analysis is a new method which has previously been applied to only measurement of carbon residues on production surfaces for pharmaceutical equipment and for water quality checking. We have applied the TOC analysis method to examine the cephradine residue. This developed and validated method offers extremely low detection capability in parts per billion (ppb), rapid sample analysis time and therefore quick turn-around of production, equipment and facilities. The method allows the measurement of extraneous materials such as process intermediates and cleaning agents, which are not possibly detected by other non specific or specific methods. TOC for cleaning validation has several advantages over specific methods. Only one method is needed for all cleaning validation analysis, the method is simpler to implement and easier to validate than chromatographic techniques and less time consuming. The method always produces a  worst-case
worst-case result, assuming that all residues are the active substance. TOC analysis demonstrated the better correlation to cleaning validation compounds in comparison to traditional analytical methods. Some qualities that make TOC a viable part of a cleaning validation include: high sensitivity, high recovery of samples, non-specific measurement and ease of use, minimal interferences and cost effectiveness. Cost savings could be attained by using cleaning validation studies. For example, by reducing a 12 h cleaning turnaround time to 6 h, per day savings on all batches could be achieved. Cephradine was chosen for cleaning, it is a cephalosporin product manufactured by most of the pharmaceutical industries. This product can cross-contaminate the other running products, which are being manufactured using the same equipment pieces like cone blender, grall mixer, encapsulation machine, blistering machine and packaging machine etc. As far as the cleaning process is concerned, cephradine has been selected due to its low water solubility and high toxicity value. This method depends on various parameters like surface type (stainless steel, glass, vinyl),9–13 and it was necessary to establish the way of addition of the drug on different surfaces and procedures to collect the sample.14,15 TOC analysis involves the oxidation of carbon and the detection of the resulting carbon dioxide. A number of different oxidation techniques exist, including photocatalytic oxidation, chemical oxidation, and high-temperature combustion.
result, assuming that all residues are the active substance. TOC analysis demonstrated the better correlation to cleaning validation compounds in comparison to traditional analytical methods. Some qualities that make TOC a viable part of a cleaning validation include: high sensitivity, high recovery of samples, non-specific measurement and ease of use, minimal interferences and cost effectiveness. Cost savings could be attained by using cleaning validation studies. For example, by reducing a 12 h cleaning turnaround time to 6 h, per day savings on all batches could be achieved. Cephradine was chosen for cleaning, it is a cephalosporin product manufactured by most of the pharmaceutical industries. This product can cross-contaminate the other running products, which are being manufactured using the same equipment pieces like cone blender, grall mixer, encapsulation machine, blistering machine and packaging machine etc. As far as the cleaning process is concerned, cephradine has been selected due to its low water solubility and high toxicity value. This method depends on various parameters like surface type (stainless steel, glass, vinyl),9–13 and it was necessary to establish the way of addition of the drug on different surfaces and procedures to collect the sample.14,15 TOC analysis involves the oxidation of carbon and the detection of the resulting carbon dioxide. A number of different oxidation techniques exist, including photocatalytic oxidation, chemical oxidation, and high-temperature combustion.
worst-case result, assuming that all residues are the active substance. TOC analysis demonstrated the better correlation to cleaning validation compounds in comparison to traditional analytical methods. Some qualities that make TOC a viable part of a cleaning validation include: high sensitivity, high recovery of samples, non-specific measurement and ease of use, minimal interferences and cost effectiveness. Cost savings could be attained by using cleaning validation studies. For example, by reducing a 12 h cleaning turnaround time to 6 h, per day savings on all batches could be achieved. Cephradine was chosen for cleaning, it is a cephalosporin product manufactured by most of the pharmaceutical industries. This product can cross-contaminate the other running products, which are being manufactured using the same equipment pieces like cone blender, grall mixer, encapsulation machine, blistering machine and packaging machine etc. As far as the cleaning process is concerned, cephradine has been selected due to its low water solubility and high toxicity value. This method depends on various parameters like surface type (stainless steel, glass, vinyl),9–13 and it was necessary to establish the way of addition of the drug on different surfaces and procedures to collect the sample.14,15 TOC analysis involves the oxidation of carbon and the detection of the resulting carbon dioxide. A number of different oxidation techniques exist, including photocatalytic oxidation, chemical oxidation, and high-temperature combustion.2. Experimental
2.1 Chemical and reagents
Cephradine reference standard was provided by Bristol Myers Squibb Pharmaceuticals, TOC grade water was prepared by Multi column distillation plant (Spirax ultra Pure System, USA). Phosphoric acid and sodium persulafte were purchased from Scharlau (Barcelona, Spain). Absorband TX762 absorbent cotton swabs were from Texwipe (Upper Saddle River, NJ).
2.2 Instrumentation and methodology
The development of this method and validation were performed on a Anatel A-2000 wide range TOC Analyzer. It measures TOC directly by adding phosphoric acid to the sample to reduce pH to approximately 2 to 3. At this low pH, any inorganic carbon that is present is liberated as CO2 into a nitrogen carrier gas and is directly measured by a non-dispersive infrared (NDIR) detector. Any remaining carbon in the sample is assumed to be TOC. A sodium persulfate oxidant is then added to the sample, and in the presence of UV radiation, the remaining carbon is oxidized to CO2. The amount of CO2 generated is then measured by NDIR to determine the amount of TOC originally present in the water.
2.3 Sample preparation
The TOC swabbing performs the swabbing procedure as follows. An aliquot of 20 ml TOC grade water into 20 ml TOC vial. Desorb a polyster tipped swab in TOC vial containing 20 ml TOC grade water. Using one side of the moistened swab, swab 100 cm2, moving from left to right and pour into TOC vial containing 20 ml TOC grade water. Using dry swab and perform additional swabbing on the same sampling area without desorbing into the water. The sealed TOC vial is vortexed for 10 s and analyzed for TOC.
3. Method validation
Linearity was tested using standard calibration curve at a concentration range of 30 to 600 ng ml−1. These standards were tested six times in agreement to ICH guidelines.16 A calibration curve was constructed and the proposed method was evaluated by its correlation coefficient and intercept value, calculated in the corresponding statistical study (ANOVA) (p < 0.05).17 The accuracy was evaluated by the recovery of cephradine (300 ng ml−1) at three different levels (150 ng ml−1, 300 ng ml−1, and 450 ng ml−1), each level tested three times. The swabbing recovery study, which involved spiking cephradine on 100 cm2 316 L stainless steel coupons, allowing the coupons to dry, recovering the cephradine with swabs, and desorbing the swabs into TOC grade water. These swabbing samples were then analyzed for TOC. Swabbing recovery included the following steps: Swabbing blank determination on ten 100 cm2 316 L stainless steel coupons was preformed. First TOC grade water was spread on all SS 316 L coupons, subsequently obtain the water sample with swab and analyzed on TOC for blank determination. Then impregnated 150 ng ml−1, 300 ng ml−1 and 350 ng ml−1 on swabs and poured into 20 ml TOC grade vials containing the same water which was used in blank preparation and vortexed for 10 s and analyzed for TOC for standard readings. For recovery studies we spread (150 ng ml−1, 300 ng ml−1 and 350 ng ml−1) of standard solution in an area of 10 × 10 cm on nine coupons. We desorbed a polyester tipped swab in TOC vial containing 20 ml TOC grade water. Using one side of the moistened swab, swab 100 cm2, moving from left to right and pour into TOC vial containing 20 ml TOC grade water. Using dry swab perform additional swabbing on the same sampling area without desorbing into the water. The sealed TOC vial is vortexed for 10 s and analyzed for TOC. According to the ICH recommendations,16 precision was considered at two levels, repeatability and intermediate precision. On this account, six-sample replicates were consecutively tested in the same equipment at a concentration of 100% of the regular analytical working value.
4. Evaluation of maximum allowable carry over
The maximum allowable carry over limit of cephradine as potential cross-contaminant was calculated through several methods.18 The total surface area of the equipment chain in direct contact with the product was accounted for in the calculations. This accounts also for the maximum daily intake of a following product and for its batch size that will be manufactured next with the same equipment. 0.1% approach was calculated by
 | ||
10 ppm approach was calculated by
MAC = 10 x S (1/A) (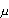 g/cm2) g/cm2) |
Where, MAC is the maximum allowable carry over residue of API permitted after cleaning, allowed into the next product; it is assumed that the total amount of residue is distributed homogenously into the following product; D the lowest daily therapeutic dose of the contaminant; S the lowest batch size of the product to follow; I the maximum daily intake of the product to follow; F the safety factor (can vary from 100 to 100 000 depending on the product nature, e.g., topical, oral or injectable preparations); A the total surface area of equipment in direct contact with the products, calculated on the basis of the assumption that all the products come into contact with all the equipment pieces of the chain.
5. Results and discussion
5.1 Method validation
 5.1.1. Accuracy. The recovery value for each concentration was calculated by comparing the blank corrected recovery mean TOC value to the blank corrected impregnated mean TOC value. The mean recovery data (mean ± R.S.D.) for each level were (115.47 ± 2.25%, 104.30 ± 1.53% and 98.10 ± 2.70% respectively, (Table 1).
5.1.1. Accuracy. The recovery value for each concentration was calculated by comparing the blank corrected recovery mean TOC value to the blank corrected impregnated mean TOC value. The mean recovery data (mean ± R.S.D.) for each level were (115.47 ± 2.25%, 104.30 ± 1.53% and 98.10 ± 2.70% respectively, (Table 1). Table 1 Accuracy
| S. No | Swabbing Blank (ppb)/A | Impregnated Sample 150 ng ml−1 (X) | Impregnated Sample 300 ng ml−1 (Y) | Impregnated Sample 450 ng ml−1 (Z) | Impregnated Sample 150 ng ml−1 - Swabbing Blank (ppb) (X-A) | Impregnated Sample 300 ng ml−1 - Swabbing Blank (ppb) (Y-A) | Impregnated Sample 450 ng ml−1 - Swabbing Blank (ppb) (Z-A) |
| 1 | 125 | 375 | 632 | 886 | 250 | 507 | 761 |
| 2 | 128 | 364 | 641 | 881 | 236 | 513 | 753 |
| 3 | 121 | 361 | 620 | 861 | 240 | 499 | 740 |
| 4 | 119 | 384 | 618 | 867 | 265 | 499 | 748 |
| 5 | 124 | 367 | 647 | 874 | 243 | 523 | 750 |
| 6 | 129 | 387 | 635 | 876 | 258 | 506 | 747 |
| 7 | 130 | 374 | 637 | 869 | 244 | 507 | 739 |
| 8 | 124 | 370 | 645 | 874 | 246 | 521 | 750 |
| 9 | 118 | 369 | 623 | 894 | 251 | 505 | 776 |
| 10 | 125 | 381 | 619 | 861 | 256 | 494 | 736 |
| Mean | 124 | 373 | 631 | 874 | 249 | 507 | 750 |
| SD | 4.0 | 8.6 | 11.0 | 10.5 | 8.9 | 9.3 | 11.7 |
| %RSD | 3.2 | 2.3 | 1.74 | 1.21 | 3.57 | 1.87 | 1.56 |
| S. No | Cephradine Recovery 150 ng ml−1 (B) | Cephradine Recovery 300 ng ml−1 (C) | Cephradine Recovery 450 ng ml−1 (D) | Cephradine Recovery 150 ng ml−1 (B) – Blank mean | Cephradine Recovery 300 ng ml−1 (C) – Blank mean | Cephradine Recovery 450 ng ml−1 (D) – Blank mean | |
| 1 | 413 | 653 | 837 | 289 | 529 | 713 | |
| 2 | 405 | 645 | 867 | 281 | 521 | 743 | |
| 3 | 428 | 661 | 875 | 294 | 537 | 751 | |
| Mean | 415 | 653 | 859 | 288 | 529 | 735 | |
| %RSD | 2.81 | 1.23 | 2.33 | 2.27 | 1.51 | 2.72 | |
| %Recovery | |||||||
| S. No | (Swabbing blank corrected recovery / Blank corrected impregnated sample Mean) X 100 | ||||||
| % Recovery 150 ng ml−1 | % Recovery 300 ng ml−1 | % Recovery 450 ng ml−1 | |||||
| 1 | 115.6 | 104.3 | 95.1 | ||||
| 2 | 112.8 | 102.7 | 99.1 | ||||
| 3 | 118.0 | 105.9 | 100.1 | ||||
| Mean | 115.47 | 104.30 | 98.10 | ||||
| %RSD | 2.25 | 1.53 | 2.70 | ||||
5.1.2 Linearity. Linearity was determined at ten levels representing from 30 to 600 ng ml−1 (10.0% to 200%) A calibration curve was constructed and the proposed method was evaluated by its correlation coefficient, slope and intercept values, which were 0.99986, 56324.18 and −5.6952 respectively. The limits of detection and quantification were 10 and 30 ng ml−1 respectively. 5.1.3 Precision. 5.1.3.1 Precision repeatability. Repeatability precision was determined by performing swabbing, which involved spiking cephradine on 316 L stainless steel coupons, recovering the cephradine with swabs, and desorbing the swabs into TOC grade water. These swabbing samples were then analyzed for TOC. Swabbing was performed with six replicates using the following cephradine concentrations: 150 ng ml−1, 300 ng ml−1 and 450 ng ml−1. The precision repeatability was performed in the same manner as in the accuracy study. The data of Table 2 shows that the average results of precision repeatability within 100 ± 10.0% of test concentrations of 150 ng ml−1, 300 ng ml−1 and 450 ng ml−1 and R.S.D. was less than 5.0% (Table 2). Table 2 Precision repeatability
| Swabbing Blank Mean (ppb) | |||||
| 115 | |||||
| Impregnated Sample 150 ng ml−1 | Impregnated Sample 150 ng ml−1 – swabbing blank mean | Impregnated Sample 300 ng ml−1 | Impregnated Sample 300 ng ml−1 – swabbing blank mean | Impregnated Sample 450 ng ml−1 | Impregnated Sample 450 ng ml−1 – swabbing blank mean |
| 415 | 300 | 649 | 534 | 890 | 775 |
| 401 | 286 | 615 | 500 | 902 | 787 |
| 398 | 283 | 641 | 526 | 867 | 752 |
| 411 | 296 | 648 | 533 | 856 | 741 |
| 387 | 272 | 610 | 495 | 892 | 777 |
| 408 | 293 | 621 | 506 | 859 | 744 |
| Mean = 288.33 | Mean = 515.67 | Mean = 762.67 | |||
| %RSD = 3.53 | %RSD = 3.37 | %RSD = 2.54 | |||
| Cephradine Recovery 150 ng ml−1 | Cephradine Recovery 150 ng ml−1 – swabbing blank mean | Cephradine Recovery 300 ng ml−1 | Cephradine Recovery 300 ng ml−1 – swabbing blank mean | Cephradine Recovery 450 ng ml−1 | Cephradine Recovery 450 ng ml−1 – swabbing blank mean |
| 435 | 320 | 655 | 540 | 905 | 790 |
| 425 | 310 | 625 | 510 | 889 | 774 |
| 405 | 290 | 612 | 497 | 914 | 799 |
| 425 | 310 | 634 | 519 | 911 | 796 |
| 401 | 286 | 627 | 512 | 896 | 781 |
| 409 | 294 | 631 | 516 | 890 | 775 |
| Mean = 301.67 | Mean = 515.67 | Mean = 785.83 | |||
| %RSD = 4.49 | %RSD = 2.74 | %RSD = 1.36 | |||
| S.No | Precision result (150 ng ml−1) | Precision result (300 ng ml−1) | Precision result (450 ng ml−1) | ||
| 1 | 106.7% | 100.9% | 101.9% | ||
| 2 | 108.3% | 101.6% | 98.3% | ||
| 3 | 102.5% | 95.4% | 106.3% | ||
| 4 | 104.7% | 97.8% | 107.4% | ||
| 5 | 105.1% | 102.7% | 100.5% | ||
| 6 | 100.3 | 101.6% | 104.2% | ||
| Mean | 104.6% | 100.04% | 103.1% | ||
| %RSD | 2.75 | 2.79 | 3.37 | ||
| Lower control limit (LCL) | 101.6% | 97.1% | 99.5% | ||
| Upper control limit (UCL) | 107.6% | 103.0% | 106.8% | ||
5.1.3.2 Precision intermediate. The second analyst carried out intermediate precision on a different day. The swabbing recoveries were performed in the same manner as in the accuracy study. The average results of precision intermediate were within ± 10.0% of test concentrations of 150 ng ml−1, 300 ng ml−1 and 450 ng ml−1 and with 92.3 – 106.8% confidence interval, which indicate a good precision. 5.1.4 Robustness. Robustness tests examine the effect that operational parameters have on the analysis results. For the determination of a method's robustness, a number of method parameters, for example, solution stability, pH, flow rate, injection volume, detection wavelength or diluent composition, are varied within a realistic range, and the quantitative influence of the variables is determined. If the influence of the parameter is within a previously specified tolerance, the parameter is said to be within the method's robustness range. In this study, only one factor was evaluated which was solution stability. The stability of swab sample taken from SS coupon was evaluated at room temperature, at intervals of 1, 24, and 48 h.19 The results obtained (mean = 107.50%, 110.25%, 91.25% respectively) revealed that samples retained a potency of 100 ± 10% as tested against freshly prepared impregnated standard solution. 5.2. %Recovery from stainless steel, vinyl and glass surfaces
Each plate (S.S. plate, Vinyl and Glass plate) was spread with variable aliquots (150 ng ml−1, 300 ng ml−1 and 350 ng/ ml) of standard solution in an area of 10 × 10 cm. Similar procedure, as used in method validation was applied to lift the residues from the surfaces. The % recoveries from each surface showed that the recovery was influenced by the type and the size of surface and not by the level of the drug spiked.
5.3. Establishing limits of cross-contamination on clean equipment
Swab sampling of areas hardest to clean was done from the equipment train used in the manufacturing and residual was found in g/swab (Table 3). The lowest obtained values were selected as limit of maximum allowable carry over (MAC) for this study.
g/swab (Table 3). The lowest obtained values were selected as limit of maximum allowable carry over (MAC) for this study.Table 3 Sample analysis from
hard to clean areas from the equipment train | Equipment name | Sampling point | Cephradine (g/ cm2) | ||
| Batch 01 | Batch 02 | Batch 03 | ||
| Cone blender | Dispensing end | 52.01 | 41.6 | 21.54 |
| Side wall | 21.21 | 17.63 | 14.72 | |
| Outlet mouth wall | 12.12 | 13.09 | 12.15 | |
| Grall mixer | Outlet | 6.9 | 9.80 | 11.6 |
| Near gasket | 61.68 | 69.6 | 38.2 | |
| Blades (chopper) | 8.9 | 68.4 | 55.7 | |
| Fitz-mill | Inside grooves | 221.5 | 324 | 142 |
| Sieve bottom | 247 | 296.2 | 315.4 | |
| Encapsulation machine | Inside punch assembly | 258.6 | 149.8 | 146.8 |
| Inside dye assembly | 44.37 | 35.6 | 69.3 | |
| Inside dye | 22.29 | 6.80 | 60.3 | |
| Blister machine | Brush | 9.6 | 2.1 | 3.8 |
| Belt | 8.4 | 6.7 | 5.9 | |
A lowest calculated value of 315 g cephradine/cm2 was obtained when the 0.1% dose limit criterion was used for the total equipment chain which was justified by the principle that an active pharmaceutical ingredient (API) at a concentration of 1/1000 of its lowest therapeutic dose will not produce any adverse effects.18
g cephradine/cm2 was obtained when the 0.1% dose limit criterion was used for the total equipment chain which was justified by the principle that an active pharmaceutical ingredient (API) at a concentration of 1/1000 of its lowest therapeutic dose will not produce any adverse effects.18 The lowest calculated value was obtained when 10 ppm acceptance criterion was applied. When less than 10 ppm of cephradine was allowed into the next manufactured product, a limit of 587 g cephradine/cm2 was determined as MAC.
g cephradine/cm2 was determined as MAC. 5.4. Assay of swab samples collected from the equipment train
Swab samples collected from different locations of the manufacturing equipment train were analyzed with the new method. For the current study it was observed that all data obtained lie within 2s of the sample mean and well below the MAC (Table 3). This gave the confidence that the manual cleaning procedures tested do provide sufficient removal of the residues from the equipment train.
6. Conclusion
A rapid and reliable TOC method for determination of residues of cephradine on pharmaceutical manufacturing plant equipment has been developed and validated. This assay technique fulfilled all the requirements to be identified as a reliable and feasible method, including accuracy, linearity, recovery and precision data. It is a non-specific and precise analytical procedure and its quick and rapid analysis allows the analysis of a large number of samples in a short period of time. Therefore, this TOC method can be used for a routine residual analysis. The level of contamination found after equipment cleaning was monitored during several consecutive runs. The results obtained confirm that the cleaning procedures used are able to remove residues from equipment surfaces well below the calculated limit of contamination.
References
| 1 | PIC/S PI 006, Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation, 2007 | |
| 2 | PDA Technical Report No. 29: Points to Consider for Cleaning Validation, PDA J. Pharm. Sci. Technol., 52, 1–23 (1998) | |
| 3 | R. C. Hwang, How to Establish an Effective Maintenance Program for Cleaning Validation, Pharm. Technol., 2000, 24, 62–67 | |
| 4 | D. A. LeBlanc, Establishing Scientifically Justified Acceptance Criteria for Cleaning Validation of Finished Drug Products, Pharm. Technol., 1998, 23, 136–148 | |
| 5 | T. M. Rossi and R. R. Ryall, Development and Validation of Analytical Methods, Pergamon Press/Elsevier, New York, 293–302 (1996) | |
| 6 | F. Laban, S.T.P. Pharma Pract., 1997, 7, 87–127 | |
| 7 | Guide to Inspections Validation of Cleaning Processes, Food and Drug Administration, Office of Regulatory Affairs, Washington DC, 1–6 (1993) | |
| 8 | PIC/S, Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation, PI 006–2, 2004 | |
| 9 | O. W. Reif, P. Solkner and J. Rupp, J. Liq. Chrom., 1996, 50, 399–410 | |
| 10 | K. D. Altria, E. Creasey and J. S. Howels, Routine capillary electrophoresis trace level determinations of pharmaceutical and detergent residues on pharmaceutical manufacturing equipment, J. Liq. Chromatogr. Relat. Technol., 1998, 21, 1093–1105 [Links]. | |
| 11 | J. Lambropoulos, G. A. Spanos and N. V. Lazaridis, Development and validation of an HPLC assay for fentanyl, alfentanil, and sufentanil in swab samples, J. Pharm. Biomed. Anal., 2000, 23, 421–428 [Links]. | |
| 12 | T. Mirza, M. Lunn, F. Keeley, R. George and J. Bodenmiller, J. Pharm. Biomed. Anal., 1999, 19, 747–756 [Links]. | |
| 13 | T. Mirza, R. George, J. Bodenmiller and S. Belanich, J. Pharm. Biomed. Anal., 1998, 16, 939–950 [Links]. | |
| 14 | C. E. Biwald and W. K. Gavlik, J. AOAC. Int., 1997, 80, 1078–1083 [Links]. | |
| 15 | A. J. Holmes and A. J. Vanderwielen, J. Liq. Chrom., 1997, 51, 149–152 | |
| 16 | ICH, Q2B Validation of analytical procedure: methodology, International Conference on Harmonisation, London, 1995 | |
| 17 | J. A. M. Pulgarín, A. Molina and M. T. Pardo, Direct determination of naftopidil by non-protected fluid room temperature phosphorescence, Analyst, 2001, 126, 234–238 [Links]. | |
| 18 | G. L. Fourman and M. V. Müllen, Determining cleaning validation acceptance limits for pharmaceutical manufacturing operations, Pharm. Technol., 1993, 17, 54–60 | |
| 19 | Validation of Chromatographic Methods, Center of Drug and Research Reviewers guidance Evaluation (1994) |


2 comments:
Our professionals use state-of-the-art software, including such modeling programs as AutoCAD, SolidWorks, and Pro Engineer to design and analyze your planned mechanisms prior to manufacturing actual prototypes read more
If you are thinking about a kit that has both the standard 18-55 lens and a second lens I suggest that you try to get the 55-300 instead of the 55-200. That extra 100 mm lets you reach out a bit further.
click here
Post a Comment